bedtools for genomics analysis
1. bedtools introduction
- bedtools is an essential command line toolkit for genomic arithmetics (sorting, finding overlap, merging, etc.), file format conversions, coverage calculations, multi-way file comparisons, and sequence manipulation analysis.
- bedtools works with several input file formats including BED, GFF, VCF, FASTA, and SAM/BAM. The records in BED files must be tab separated.
- BED (Browser Extensible Data) file contains 12 columns including chr, start, end, name, score, strand, thickstart, thickend, itemRgb, blockCount, blockSizes, and blockStarts. But, bedtools also supports the first three columns (chr, start, end) for common purpose utilities. Check the description of each column here.
- There are various types of BED files (e.g. BED3, BED6, BED12, etc.) based on the presence of a number of columns. The most common BED3 format contains the first three columns and the BED12 format contains all 12 columns.
- The genomic coordinates in BED files are 0-based (start position) as opposed to GFF and VCF which contains the coordinates in 1-based format. bedtools adjusts the coordinates accordingly if the input is in GFF or VCF file formats.
2. How to install bedtools
bedtools is available only for Linux and Mac. The easiest way to install bedtools using package managers such as
apt-get (Ubuntu) and homebrew (Mac OS)
Learn more about Linux commands for Bioinformatics
2.1 Install on Linux (Ubuntu)
# using package manager
sudo apt update
sudo apt-get install bedtools
# using static binary
wget https://github.com/arq5x/bedtools2/releases/download/v2.30.0/bedtools.static.binary
mv bedtools.static.binary bedtools
chmod a+x bedtools
export PATH=/home/renesh/lin_proj/software:$PATH
2.2 Install on Mac
brew tap homebrew/science
brew install bedtools
# MacPorts
port install bedtools
2.3 Check version
bedtools --version
# output
bedtools v2.30.0
3. bedtools common utilities
3.1 Compare two BED files for overlapping coordinates
bedtools intersect (aka intersectBed) helps you to identify overlapping and non-overlapping genomic features in two BED, GFF, VCF, or
BAM files.
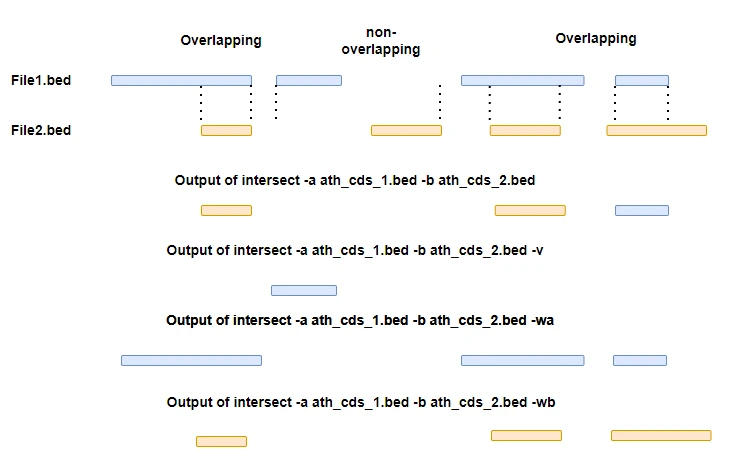
Find overlapping features between two BED files,
Download files: ath_cds_1.bed, ath_cds_2.bed and ath_cds_3.bed
bedtools intersect -a ath_cds_1.bed -b ath_cds_2.bed
# output
Chr1 5300 5326
Chr1 5439 5630
Reports all genomic coordinates from -a that overlapped with -b file,
bedtools intersect -a ath_cds_1.bed -b ath_cds_2.bed -wa
# output
Chr1 5174 5326
Chr1 5439 5630
Report all non-overlapped genomic coordinates from -a file,
bedtools intersect -a ath_cds_1.bed -b ath_cds_2.bed -v
# output
Chr1 3996 4276
Chr1 4486 4605
Chr1 4706 5095
Report all overlapped and non-overlapped genomic coordinates from -a file (like left outer join)
bedtools intersect -a ath_cds_1.bed -b ath_cds_2.bed -loj
# output
Chr1 3996 4276 . -1 -1
Chr1 4486 4605 . -1 -1
Chr1 4706 5095 . -1 -1
Chr1 5174 5326 Chr1 5300 5326
Chr1 5439 5630 Chr1 5439 5630
3.2 Compare multiple files for overlapping coordinates
bedtools multiinter (aka multiIntersectBed)) helps you to identify overlapping and non-overlapping genomic features
in multiple BED, GFF, or VCF files. The input files must be sorted by chromosome and start positions.
Find overlapping features between three BED files,
bedtools multiinter -i ath_cds_1.bed ath_cds_2.bed ath_cds_3.bed -header
# output
chrom start end num list ath_cds_1.bed ath_cds_2.bed ath_cds_3.bed
Chr1 3996 4276 2 1,3 1 0 1 # overlapped in first and third bed files
Chr1 4486 4605 1 1 1 0 0
Chr1 4706 5095 2 1,3 1 0 1 # overlapped in first and third bed files
Chr1 5174 5300 1 1 1 0 0
Chr1 5300 5326 3 1,2,3 1 1 1 # overlapped in all three bed files
Chr1 5439 5630 2 1,2 1 1 0 # overlapped in first two bed files
Chr1 7942 7987 1 2 0 1 0
Chr1 8236 8325 1 2 0 1 0
Chr1 8417 8464 1 2 0 1 0
Chr2 5439 5630 1 3 0 0 1
3.3 Calculate the depth and breadth of coverage
bedtools coverage utility helps you to calculate both depth and breadth of coverage
between features between two BED, GFF, or VCF files. Basically, it is similar to intersect but give detailed reports
on coverage analysis such as how many features from -b file overlapped with -a file (depth of coverage), how
many bases are covered (breadth of coverage), and their covered fractions (0 to 1 scale).
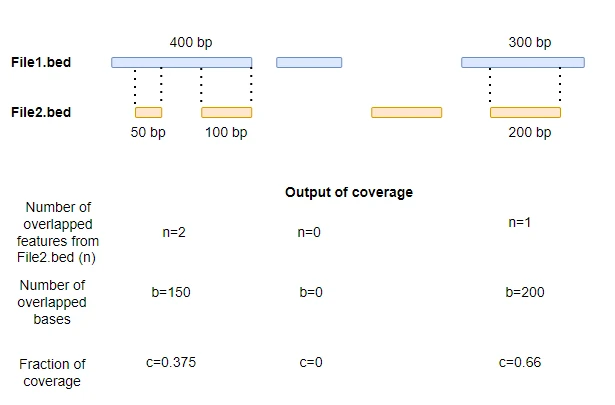
By default, all features from -a file are reported in output. After each column in -a file, the output contains columns
for number of overlapped features from -b file, the number of overlapped bases from -b file, length of feature
from -a file, and their covered fractions (0 to 1 scale).
bedtools coverage -a ath_cds_1.bed -b ath_cds_2.bed
Chr1 3996 4276 0 0 280 0.0000000
Chr1 4486 4605 0 0 119 0.0000000
Chr1 4706 5095 0 0 389 0.0000000
Chr1 5174 5326 1 26 152 0.1710526
Chr1 5439 5630 1 191 191 1.0000000
For example, the feature (Chr1: 5174, 5326) from ath_cds_1.bed overlaps with one feature from ath_cds_2.bed, and it’s 17.10% of bases are covered.
3.4 Extract the sequence in FASTA format from BED file
bedtools getfasta (aka fastaFromBed) utility is useful to extract the sequence in FASTA format using the feature
coordinates provided in BED, GFF, or VCF files.
bedtools getfasta -fi Athaliana_167_TAIR10.fa -bed at_cds.bed -fo at_cds.fasta
The output from above command will be saved in at_cds.fasta file
head at_cds.fasta
# output
>Chr1:3631-5899
AATTATTAGATATACCAAACCAGAGAAAACAAATACATAATCGGAGAAATACAGATTACAGAGAGCGAGAGAGATCGACGGCGAAGCTCTTTACCCGGAAACCATTGAAATCGGACG
GTTTAGTGAAAATGGAGGATCAAGTTGGGTTTGGGTTCCGTCCGAACGACGAGGAGCTCGTTGGTCACTATCTCCGTAACAAAATCGAAGGAAACACTAGCCGCGACGTTGAAGTAG
CCATCAGCGAGGTCAACATCTGTAGCTACGATCCTTGGAACTTGCGCTGTAAGTTCCGAATTTTCTGAATTTCATTTGCAAGTAATCGATTTAGGTTTTTGATTTTAGGGTTTTTTT
T
(sequence is truncated for representation)
Note: The input genome FASTA must be indexed. You can index the genome using
samtools faidxutility
Learn more about how to extract the DNA sequences from reference FASTA using bedtools getfasta
3.5 File format conversions
bedtools bedtobam (aka bedToBam), bamtobed (aka bamToBed), and bamtofastq are commonly used functions for
file format conversions.
Convert BED to BAM format,
Download files: ath_cds.bed
bedtools bedtobam -i ath_cds.bed -g ath_chr1.sizes > ath_cds.bam
# view BAM records
samtools view ath_cds.bam | head -2
# output
cds1 0 Chr1 3997 255 280M * 0 0 * *
cds2 0 Chr1 4487 255 119M * 0 0 * *
Note: BED file should also contain the 4th name column. In addition, bedtools uses genome file in chromosome coordinates format and not in FASTA format. The genome file should contain chr and size columns separated by a tab.
Convert BAM to BED format,
bedtools bamtobed -i ath_cds.bam > ath_cds_con.bed
# view BED records
head -2 ath_cds_con.bed
# output
Chr1 3996 4276 cds1 255 +
Chr1 4486 4605 cds2 255 +
Convert BAM to FASTQ format,
bedtools bamtofastq -i sample.bam -fq sample.fastq
# BAM with paired-end reads
bedtools bamtofastq -i sample.bam -fq sample_1.fastq -fq2 sample_2.fastq
# create interleaved FASTQ file
bedtools bamtofastq -i sample.bam -fq /dev/stdout -fq2 /dev/stdout > sample_interleaved.fastq
3.6 Calculate GC and AT content
bedtools nuc (aka nucBed) can be used for calculating the GC content for genomic features available in BED, GFF, or
VCF files. It requires sequence (FASTA) and genomic features (BED/GFF/VCF).
bedtools nuc -fi Athaliana_167_TAIR10.fa -bed Athaliana_167_gene.gff3 > gc_content.txt
Once you run the above command successfully, the gc_content.txt file contains several records including GC content (11th),
AT content (10th column), and other sequence statistics. The output columns are appended to the GFF columns. You can
see all detailed output information by using bedtools nuc -h
cut -f1-11 gc_content.txt | head
# output
#1_usercol 2_usercol 3_usercol 4_usercol 5_usercol 6_usercol 7_usercol 8_usercol 9_usercol 10_pct_at 11_pct_gc
Chr1 phytozome9_0 gene 3631 5899 . + . ID=AT1G01010;Name=AT1G01010 0.613927 0.386073
Chr1 phytozome9_0 mRNA 3631 5899 . + . ID=PAC:19656964;Name=AT1G01010.1;pacid=19656964;longest=1;Parent=AT1G01010 0.613927 0.386073
Chr1 phytozome9_0 five_prime_UTR 3631 3759 . + . ID=PAC:19656964.five_prime_UTR.1;Parent=PAC:19656964;pacid=19656964 0.612403 0.387597
Chr1 phytozome9_0 CDS 3760 3913 . + 0 ID=PAC:19656964.CDS.1;Parent=PAC:19656964;pacid=19656964 0.487013 0.512987
Chr1 phytozome9_0 CDS 3996 4276 . + 2 ID=PAC:19656964.CDS.2;Parent=PAC:19656964;pacid=19656964 0.551601 0.448399
Chr1 phytozome9_0 CDS 4486 4605 . + 0 ID=PAC:19656964.CDS.3;Parent=PAC:19656964;pacid=19656964 0.541667 0.458333
Chr1 phytozome9_0 CDS 4706 5095 . + 0 ID=PAC:19656964.CDS.4;Parent=PAC:19656964;pacid=19656964 0.582051 0.417949
Chr1 phytozome9_0 CDS 5174 5326 . + 0 ID=PAC:19656964.CDS.5;Parent=PAC:19656964;pacid=19656964 0.588235 0.411765
Chr1 phytozome9_0 CDS 5439 5630 . + 0 ID=PAC:19656964.CDS.6;Parent=PAC:19656964;pacid=19656964 0.567708 0.432292
The %GC and %AT content for AT1G01010 gene is 38.60% and 61.37% respectively.
3.7 Sort BED files
Sort BED file based on chromosome name and start position,
Download files: ath_cds_4.bed
bedtools sort -i ath_cds_4.bed
# output
Chr1 3996 4276
Chr1 4706 5095
Chr2 5439 5630
Sort BED file based on feature size,
# ASC order of feature size
bedtools sort -i ath_cds_4.bed -sizeA
# output
Chr2 5439 5630
Chr1 3996 4276
Chr1 4706 5095
# DESC order of feature size
bedtools sort -i ath_cds_4.bed -sizeD
# output
Chr1 4706 5095
Chr1 3996 4276
Chr2 5439 5630
3.8 Split BED file by chromosome
bedtools does not have function to split the BED file by a choromosme. But you can use bioinfokit.HtsAna.split_bed
package to split the BED file by chromosome names.
from bioinfokit.analys import HtsAna
HtsAna.split_bed('ath_cds_1.bed')
The split files by each chromosome name will be saved in the same directory
3.8 Merge intervals in a BED file
If there are any overlapping or nearby coordinate intervals in the BED (or VCF or GFF) file, you can merge them into a single interval.
Download example BED file: ath_cds_5.bed
For example, merge a overlapping coordinate intervals,
# use -s parameter if you want merge features from same strand
bedtools merge -i ath_cds_5.bed
# output
Chr1 3996 4276
Chr1 4000 4276
Chr1 4486 4605
Chr1 4706 5095
Chr1 5174 5326
Chr1 5439 5630
Chr1 5670 6680
Merge coordinate intervals with a specified distance,
# merge inbtervals when they are within 60 bp apart
# use -s parameter if you want merge features from same strand
bedtools merge -d 60 -i ath_cds_5.bed
# output
Chr1 3996 4276
Chr1 4486 4605
Chr1 4706 5095
Chr1 5174 5326
Chr1 5439 6680
Enhance your skills with courses on genomics and bioinformatics
- Genomic Data Science Specialization
- Biology Meets Programming: Bioinformatics for Beginners
- Command Line Tools for Genomic Data Science
- Data Science: Foundations using R Specialization
- Python for Genomic Data Science
- Bioinformatics Specialization
References
If you have any questions, comments or recommendations, please email me at reneshbe@gmail.com
This work is licensed under a Creative Commons Attribution 4.0 International License
Some of the links on this page may be affiliate links, which means we may get an affiliate commission on a valid purchase. The retailer will pay the commission at no additional cost to you.

