Mastering RNA-seq Read Alignment With STAR

Page content
Background
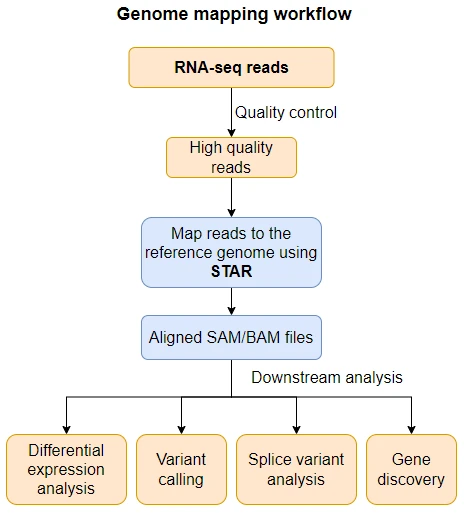
- The alignment (mapping) of high-throughput sequencing reads to a reference genome is a critical step in RNA-seq and DNA-seq data analysis. The mapping of sequence reads to reference genome helps in gene discovery, gene quantification, splice variant (alternative splicing) analysis, variant calling, and identifying chimeric (fusion) genes.
- Spliced Transcripts Alignment to a Reference (STAR) is a highly accurate and ultra-fast splice-aware aligner for aligning RNA-seq reads to the reference genome sequences. STAR is the recommended aligner for mapping RNA-seq data.
- STAR has a better mapping rate as compared to other popular splice-aware RNA-seq aligners such as HISAT2 and TopHat2. STAR is faster than TopHat2. The drawback of STAR is that it is RAM (memory) intensive aligner and may require high-end computers for analysis. The aligning speed for STAR can vary based on the available memory.
- STAR has high precision in identifying the canonical and non-canonical (novel) splice junctions. STAR can also detect the chimeric (fusion) transcripts. In addition to mapping short reads (e.g. ≤ 200 bp), STAR can accurately map long reads (e.g. several Kbp of reads generated from PacBio or Ion Torrent).
- In addition, STAR also has better sensitivity in variant detections (SNPs and INDELs) as compared to TopHat2. Hence, STAR is used in GATK best practice workflow for indetifying short variants from RNA-seq data.
Getting started with STAR
Computational requirements for STAR
As RNA-seq and DNA-seq experiments generate millions of sequence reads, computers with sufficient memory and disk space are required to run the analyses.
STAR can run smoothly on high-performance computers (HPCs). If you do not have access to HPC, you should have computers that meet the following specifications
- Linux or Mac OS
- Sufficient RAM (recommended 32 GB for human size genome). It can be less for smaller genomes.
- Hard drive with sufficient disk space based on the input file sizes (recommended 100 to 500 GB)
- Multiple processors (cores) for parallel computation (e.g. Xeon processor)
Install STAR
If you have not previously installed STAR, you can download the pre-compiled binaries for STAR. Alternatively, you can also install it from the source by following the installation instructions.
# for Linux
git clone https://github.com/alexdobin/STAR.git
# add binaries to path using export path or editing ~/.bashrc file
export PATH=$PATH:/home/ren/software/STAR/bin/Linux_x86_64
# verify the binaries added to the system path
which STAR
# output
/home/ren/software/STAR/bin/Linux_x86_64/STAR
Once, the binaries successfully added to PATH, you should able to see the complete STAR usage by typing the STAR -h command.
Mapping reads with STAR
STAR workflow
Mapping reads to a reference genome using STAR includes two steps,
- Building reference genome index using genome (FASTA) and annotation (GTF/GFF3) files
- Mapping reads (FASTQ or FASTA) to indexed genome
Building genome indices
In this tutorial, I am using the Arabidopsis thaliana genome for building the index and mapping the reads to the genome.
STAR uses the genome file (FASTA) and gene annotation file (GTF or GFF3) to create the genome indices. The gene annotation file is needed for creating the known splice junctions to improve the accuracy of the genome mapping. Gene annotation file is optional, but it is highly recommended if it is available (Note: you can also provide an annotation file during the mapping step).
# STAR version=2.7.10b
# build genome index using GTF file
STAR --runThreadN 12 \
--runMode genomeGenerate \
--genomeDir ath_star_index \
--genomeFastaFiles Athaliana_TAIR10.fasta \
--sjdbGTFfile Athaliana_gene.gtf \
--sjdbOverhang 149
STAR Parameters description for building genome indices,
| Parameter | Description |
|---|---|
--runThreadN |
Number of threads (processors) for mapping reads to a genome |
--runMode |
run mode for STAR. genomeGenerate mode builds genome index |
--genomeDir |
PATH to the directory where genome indices will be stored |
--genomeFastaFiles |
reference genome file in FASTA format. You can also provide multiple genome files (separated by space) for index building. |
--sjdbGTFfile |
GTF file for gene annotation. This is an optional parameter |
--sjdbOverhang |
length of reads around the splice junctions. The ideal values should read length - 1 (or max read length - 1). For example, if your read length is 150, the value should be 149. In most cases, the default value of 100 also works. |
If you do not have GTF annotation file, you could also use a GFF3 file for building the genome indices (optionally, you can also use GFF3 to GTF file converter).
If you use a GFF3 file, you need to add an additional parameter --sjdbGTFtagExonParentTranscript Parent for defining the
parent-child relationship.
# build genome index using GFF3 file
STAR --runThreadN 12 \
--runMode genomeGenerate \
--genomeDir ath_star_index \
--genomeFastaFiles Athaliana_TAIR10.fasta \
--sjdbGTFfile Athaliana_gene.gff3 \
--sjdbGTFtagExonParentTranscript Parent \
--sjdbOverhang 149
Mapping reads to genome
Once the genome indices are created, the single and paired-end RNA-seq reads can be mapped to the reference genome using STAR (default 1-pass mapping).
For single-end reads,
# STAR version=2.7.10b
# map single end reads to genome
STAR --runThreadN 12 \
--readFilesIn ath_seed_sample.fastq \
--genomeDir ath_star_index \
--outSAMtype BAM SortedByCoordinate \
--outFileNamePrefix seed_sample \
--outSAMunmapped Within
For paired-end reads,
# map paired-end reads to genome
STAR --runThreadN 12 \
--readFilesIn ath_sample_read1.fastq ath_sample_read2.fastq \
--genomeDir ath_star_index \
--outSAMtype BAM SortedByCoordinate \
--outFileNamePrefix seed_sample \
--outSAMunmapped Within
If the read files are gzip compressed (*.fastq.gz), you can add an additional --readFilesCommand zcat or
--readFilesCommand gunzip -c parameter to the above mapping command.
STAR Parameters description for mapping reads to genome,
| Parameter | Description |
|---|---|
--runThreadN |
Number of threads (processors) for mapping reads to genome |
--readFilesIn |
Read files for mapping to the genome. In case of paired-end reads, provide read1 and read2 files. If there are multiple samples, separate files by a comma. For example, for paired-end reads, --readFilesIn S1read1.fastq,S2read1.fastq S1read2.fastq,S2read2.fastq |
--genomeDir |
PATH to the directory containing built genome indices |
--outSAMtype BAM SortedByCoordinate |
Output coordinate sorted BAM file which is useful for many downstream analyses. This is optional. |
--outSAMunmapped Within |
Output unmapped reads from the main SAM file in SAM format. This is optional |
--outFileNamePrefix |
Provide output file prefix name |
If your study goal is to identify novel splice junctions (e.g. for differential splicing analysis), it is recommended to use 2-pass mapping by re-building the genome indices using splice junctions (see
SJ.out.tabfrom output files) obtained from 1-pass. Read my article on how to use STAR for 2-pass mapping.
STAR Output files
After successful mapping, the STAR generates several output files (based on the input command) for analyzing the mapping data. The following files are output from the paired-end reads mapped to the Arabidopsis genome (command is above).
| Parameter | Description |
|---|---|
seed_sampleAligned.sortedByCoord.out.bam |
Alignment in BAM format (sorted by coordinate) |
seed_sampleLog.final.out |
Alignment summary statistics such as uniquely mapped reads, percent mapping, number of unmapped reads, etc. |
seed_sampleLog.out |
Alignment log for commands and parameters (useful in troubleshooting) |
seed_sampleLog.progress.out |
Alignment progress report (e.g. number of reads processed during particular span of time, mapped and unmapped reads, etc.) |
seed_sampleSJ.out.tab |
Filtered splice junctions found during the mapping stage |
Now, let’s check the alignment summary from these results files,
The Alignment summary statistics file (seed_sampleLog.final.out) gives detailed statistics such as the number of
input reads, average read lengths, uniquely mapped reads counts and percentages, spliced reads, chimeric reads, and a number of
unmapped reads.
less seed_sampleLog.final.out
# output
Started job on | Nov 21 20:35:58
Started mapping on | Nov 21 20:36:53
Finished on | Nov 22 08:30:49
Mapping speed, Million of reads per hour | 1.00
Number of input reads | 11889751
Average input read length | 202
UNIQUE READS:
Uniquely mapped reads number | 10757605
Uniquely mapped reads % | 90.48%
Average mapped length | 200.74
Number of splices: Total | 7206200
Number of splices: Annotated (sjdb) | 7121304
Number of splices: GT/AG | 7126695
Number of splices: GC/AG | 52558
Number of splices: AT/AC | 7593
Number of splices: Non-canonical | 19354
Mismatch rate per base, % | 0.65%
Deletion rate per base | 0.04%
Deletion average length | 1.79
Insertion rate per base | 0.02%
Insertion average length | 1.83
MULTI-MAPPING READS:
Number of reads mapped to multiple loci | 779032
% of reads mapped to multiple loci | 6.55%
Number of reads mapped to too many loci | 262
% of reads mapped to too many loci | 0.00%
UNMAPPED READS:
Number of reads unmapped: too many mismatches | 0
% of reads unmapped: too many mismatches | 0.00%
Number of reads unmapped: too short | 352683
% of reads unmapped: too short | 2.97%
Number of reads unmapped: other | 169
% of reads unmapped: other | 0.00%
CHIMERIC READS:
Number of chimeric reads | 0
% of chimeric reads | 0.00%
You can also view the read alignments using the samtools. You need to have samtools
installed for viewing BAM files. I have written a detailed article on how to install samtools
and view the BAM files.
# view first few alignment of BAM files
samtools view seed_sampleAligned.sortedByCoord.out.bam | head -n2
# output
SRR22309490.1539086 163 Chr1 3657 255 101M = 3708 152 AAAACAAATACATAATCGGAGAAATACAGATTACAGAGAGCGAGAGAGATCGACGGCGAAGCTCTTTACCCGGAAACCATTGAAATCGGACGGTTTAGTGA AAFFFJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJFJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJAJJJJFF NH:i:1 HI:i:1 AS:i:200 nM:i:0
SRR22309490.1539086 83 Chr1 3708 255 101M = 3657 -152 GACGGCGAAGCTCTTTACCCGGAAACCATTGAAATCGGACGGTTTAGTGAAAATGGAGGATCAAGTTGGGTTTGGGTTCCGTCCGAACGACGAGGAGCTCG JJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJFFFAA NH:i:1 HI:i:1 AS:i:200 nM:i:0
Read my article on how to use STAR for 2-pass mapping
Enhance your skills with courses on genomics and bioinformatics
- Genomic Data Science Specialization
- Biology Meets Programming: Bioinformatics for Beginners
- Python for Genomic Data Science
- Bioinformatics Specialization
- Command Line Tools for Genomic Data Science
- Introduction to Genomic Technologies
References
- Schaarschmidt S, Fischer A, Zuther E, Hincha DK. Evaluation of seven different RNA-seq alignment tools based on experimental data from the model plant Arabidopsis thaliana. International journal of molecular sciences. 2020 Mar 3;21(5):1720.
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013 Jan 1;29(1):15-21.
- Baruzzo G, Hayer KE, Kim EJ, Di Camillo B, FitzGerald GA, Grant GR. Simulation-based comprehensive benchmarking of RNA-seq aligners. Nature methods. 2017 Feb;14(2):135-9.
If you have any questions, comments or recommendations, please email me at reneshbe@gmail.com
This work is licensed under a Creative Commons Attribution 4.0 International License
Some of the links on this page may be affiliate links, which means we may get an affiliate commission on a valid purchase. The retailer will pay the commission at no additional cost to you.

