HISAT2: Fast Aligner for NGS Data (complete Tutorial)
What is HISAT2?
HISAT2 (hierarchical indexing for spliced alignment of transcripts 2) is a fast and sensitive splice-aware sequence alignment tool for aligning NGS generated DNA and RNA reads to the reference genomes.
For example, in transcriptomics (RNA-seq) analysis, HISAT2 can be used for aligning RNA-seq reads to the genome.
The aligned reads (HISAT2 output) are further used in various bioinformatics downstream analyses such as gene quantification, transcript assembly, and identifying differentially expressed genes and transcripts.
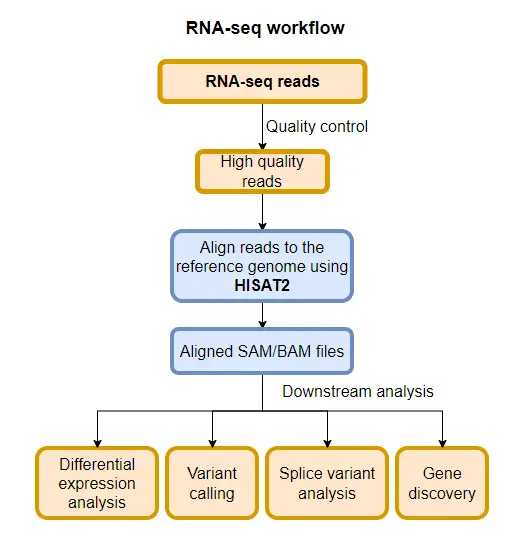
As compared to STAR, HISAT2 is a memory-efficient aligner that requires relatively low (~6.7 GB) memory for mapping NGS reads to the human genome. Further, HISAT2 is a faster aligner as compared to other competitive aligners such as STAR and Bowtie2.
Besides, HISAT2 is known to have a higher accuracy for aligning the NGS reads containing SNPs.
Tip: HISAT2 is a successor of HISAT and TopHat2 and it is recommended to use HISAT2 over HISAT and TopHat2
Getting started with HISAT2
This tutorial explains computational requirements for HISAT2, how to download and install HISAT2, and how to align NSG reads to the reference genome using HISAT2.
Computational requirements for HISAT2
Though HISAT2 is a memory-efficient aligner, your computer need to have the following specifications to run HISAT2.
- 64 bit computer with either Linux or Mac OS
- At least 8 GB RAM
- Sufficient space on hard drive (recommended 100 to 500 GB based on the input file and genome sizes)
- Multiple cores for parallel computing (e.g. Xeon processor)
How to download and install HISAT2
If you have not installed the HISAT2 on your computer, you can install it on Linux (e.g. Ubuntu) or Mac OS using the following two preferred methods.
-
Download HISAT2 executables
Download the HISAT2 v2.2.1 executables and add them to the system path
Tip: If you download executables, you should add the HISAT2 directory to the system PATH. This will help to run
hisat2commands without specifying the complete path to the HISAT2 directory.
- Install using conda
conda install -c bioconda hisat2
Alignment with HISAT2
The HISAT2 workflow for alignment of NGS reads with HISAT2 to the reference genome involves two steps,
- Building a genome index
- Aligning the NGS reads to the indexed genome
Building genome index
In this tutorial, I will use the example of Arabidopsis thaliana genome for building the genome index using HISAT2 (v2.2.1). You can download the Arabidopsis thaliana genome sequence (FASTA format) from TAIR.
You can use hisat2-build build command for building genome index as follows,
hisat2-build -p 6 -f Athaliana_TAIR10.fasta Athaliana
hisat2-build will take ~5 min to create genome index. Time to create genome index is depends on the genome size.
Where,
-p: numbers of threads for parallel computation-f: reference genome is in FASTA format
The Athalina at the end of the command is a basename for the genome index. Once the genome index is created, the
basename will be appended to the indexed genome.
Note: HISAT2 is capable of generating a genome index for any genome size. If the genome size is small (< 4 B nucleotides), it creates a small index (.ht2 extension index files). For larger genome sizes, HISAT2 generates large index (.ht2l extension index files).
Optionally, HISAT2 also uses the gene annotation file (GTF format) to extract the exons and splice sites. This is recommended to use the gene annotation for RNA-seq data analysis.
# extract exons
hisat2_extract_exons.py Athaliana_gene.gtf > Athaliana.exon
# extract splice sites
hisat2_extract_splice_sites.py Athaliana_gene.gtf > Athaliana.ss
Now, build the genome index,
hisat2-build -p 6 --exon Athaliana.exon --ss Athaliana.ss Athaliana_TAIR10.fasta Athaliana
Tip: To build a genome index with gene annotation, the
hisat2-buildwill require larger RAM (200 GB for human size genome) and may take a longer time to run (~1 hour for human genome). As an alternative, you can provide the splice sites file during the alignment stage (hisat2) using the--known-splicesite-infileparameter.
Aligning reads to genome
HISAT2 can be used to align the single and paired-end NGS reads to the reference genome once the genome indices are created.
If you want to align single-end reads, then you should pass -U parameter to hisat2,
hisat2 --phred33 --dta -x Athaliana -U ath_seed_sample_R1.fastq -S alignment.sam
Where,
--phred33: Sequence quality score. Most Illumina sequencers generate sequences in PHRED33 format.--dta: Use this option to output alignments suitable for transcriptome assembly. Mostly used for RNA-seq data analysis.-S: Output alignment to file (SAM format) instead of standard output-x: basename for indexed genome
You can read the HISAT2 manual for controlling other parameters such as output options, mismatch penalties, and input options.
If you want to align paired-end reads, then you should pass -1 and -2 parameter to hisat2,
hisat2 --phred33 --dta -x Athaliana -1 ath_seed_sample_R1.fastq -2 ath_seed_sample_R2.fastq -S alignment.sam
hisat2 will take ~15 min to align the paired-end reads to genome. This can vary based on genome and input file size.
If you have multiple paired-end files (from different samples), you can align them to genome at once,
hisat2 --phred33 --dta -x Athaliana -1 ath_seed_sample_R1.fastq,ath_root_sample_R1.fastq -2 ath_seed_sample_R2.fastq,ath_root_sample_R2.fastq -S alignment.sam
If you have skipped the gene annotation (splice sites and exons) for building genome index (due to memory restrictions),
you can provide the gene annotation information such as splice sites during the alignment stage using --known-splicesite-infile parameter.
hisat2 --phred33 --dta -x Athaliana --known-splicesite-infile Athaliana.ss -1 ath_seed_sample_R1.fastq -2
ath_seed_sample_R2.fastq -S alignment.sam
HISAT2 alignment output
The HISAT2 writes the alignment in SAM format. You can view and read the SAM files (similar to general text files).
But, most of the downstream bioinformatics analysis such as gene quantification, assembly, differential expression analysis, etc. requires alignment file in BAM format (binary file). BAM file is also useful for storage purposes as it takes less space than SAM file.
Please read this article on how to convert a SAM file into a BAM file.
In addition to alignment in SAM format, hisat2 also writes the summary statistics of alignment on the console as a standard error
message.
The following is an example of alignment summary statistics obtained from aligning the paired-end reads.
11889751 reads; of these:
11889751 (100.00%) were paired; of these:
1298966 (10.93%) aligned concordantly 0 times
9865016 (82.97%) aligned concordantly exactly 1 time
725769 (6.10%) aligned concordantly >1 times
----
1298966 pairs aligned concordantly 0 times; of these:
74231 (5.71%) aligned discordantly 1 time
----
1224735 pairs aligned 0 times concordantly or discordantly; of these:
2449470 mates make up the pairs; of these:
1594292 (65.09%) aligned 0 times
816748 (33.34%) aligned exactly 1 time
38430 (1.57%) aligned >1 times
93.30% overall alignment rate
The alignment summary statistics show that the overall alignment rate is 93.30%.
In alignment summary statistics, the indentations are useful to break down the total. For example, 11889751 (total paired reads) is a total of 1298966 (aligned concordantly 0 times), 9865016 (aligned concordantly exactly 1 time), and 725769 (aligned concordantly >1 times).
Enhance your skills with courses on genomics and bioinformatics
- Genomic Data Science Specialization
- Biology Meets Programming: Bioinformatics for Beginners
- Python for Genomic Data Science
- Bioinformatics Specialization
- Command Line Tools for Genomic Data Science
- Introduction to Genomic Technologies
References
- Musich R, Cadle-Davidson L, Osier MV. Comparison of short-read sequence aligners indicates strengths and weaknesses for biologists to consider. Frontiers in Plant Science. 2021:692.
- Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nature biotechnology. 2019 Aug;37(8):907-15.
This work is licensed under a Creative Commons Attribution 4.0 International License
Some of the links on this page may be affiliate links, which means we may get an affiliate commission on a valid purchase. The retailer will pay the commission at no additional cost to you.

