Sequencing coverage and breadth of coverage
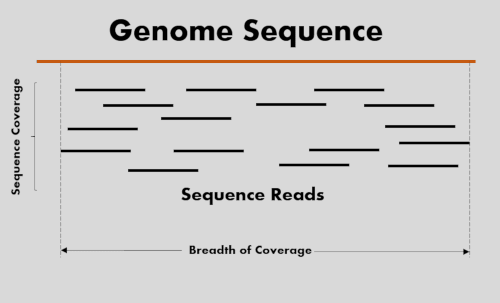
Coverage depth and breadth of coverage
- Sequencing coverage depth determines the number of times sequenced nucleotide bases covered the target genome. For example, if genome size is 100 Mbp and you have sequenced 5 M reads of 100 bp size, then sequencing coverage at genome level would be 5X.
- The breadth of coverage refers to the percentage of genome bases sequenced at a given sequencing depth. For example, if 95% of the genome is covered by sequencing at a certain depth.
How to calculate sequencing coverage
- Sequencing coverage is calculated based on the type of sequencing. For RNA-seq applications, coverage is calculated based on the transcriptome size and for genome sequencing applications, coverage is calculated based on the genome size
- Generally in RNA-seq experiments, the read depth (number of reads per sample) is used instead of coverage. High read depth is necessary to identify genes with low expressions. The typical read depth RNA-seq experiment to study gene expression ranges from 5 to 25 M reads per sample.
- Calculating sequencing coverage based on raw sequence reads will give you rough estimates as some of these raw reads may have contamination (adapter, primer, duplicates or low-quality bases) or may not map to genome. In such cases, you can consider genome mapped data for estimating the coverage.
We will use bioinfokit (v0.9.7 or later)
Check bioinfokit documentation for installation and documentation
# you can use interactive python interpreter, jupyter notebook, spyder or python code
# I am using interactive python interpreter (Python 3.7)
# go to a directory where fastq files are saved. Make sure fastq file is uncompressed.
# this will give sequencing coverage per sample
>>> from bioinfokit.analys import fastq
>>> fastq.seqcov(file="fastq_file", gs="genome size in Mbp")
In addition, you can also use samtools to calculate the coverage depth and breadth of coverage
On-target rate
- The On-target rate is commonly used terminology to measure sensitivity (% target bases in sequence reads) and specificity (% of sequence reads on target site) in targeted enrichment NGS methods such as whole exome sequencing (WES).
- On-target rate refers to how many nucleotide bases or reads are covered at the target site. On-target rate is determined by percent on-target bases (percentages of nucleotide bases mapped to the target region of the genome) and percent on-target reads (percentages of sequencing reads that covers the target region of the genome)
Uniformity of coverage
- Uniformity of coverage or coverage uniformity is a widely used method to assess the quality of the sequencing data in targeted enrichment NGS methods such as whole exome sequencing (WES). Uniformity of coverage is important quality metric in variance detection.
- Uniformity of coverage describes how sequencing reads are distributed across the targeted region in the genome. It checks the equality of sequence reads distribution across targeted regions. Ideally, all targeted regions should have sequenced reads with desired mean coverage, but this does not happen in real experiments. Some targets get higher coverage, while other targets get lower coverage. The reads also map to the off-target sites.
- Uniformity of coverage can be assessed by the fold-80 base penalty and percentages of target bases covered at least 0.2X of mean coverage methods.
- Fold-80 base penalty calculates the amount of additional sequencing (fold coverage change for non-zero reads) needed to achieve the observed mean coverage by 80% of the target bases.
- The perfect uniformity of coverage is obtained when the fold-80 base penalty is 1 (100% on-target rate). The lower value of the fold-80 base penalty represents lower variability and better uniformity (cost-effective sequencing).
- Fold-80 base penalty metric can be calculated from Picard CollectHsMetrics tool.
Recommended sequence coverage for sequencing applications
| Sequencing applications | Recommended Coverage |
|---|---|
| Whole genome sequencing (WGS) | 15X to 60X |
| Whole exome sequencing (WES) | 100X |
| RNA sequencing (RNA-seq) | 5 to 100 M reads per sample depending on target study |
| ChIP-Seq | 100X |
| Whole genome sequencing (WGS) for de novo assembly (PacBio HiFi reads) | 10X-15X per haplotype |
| Whole genome sequencing (WGS) for variant detection (PacBio HiFi reads) | ≥ 15X (for human genome) |
Source: Illumina and genohub
Enhance your skills with courses on genomics and bioinformatics
- Genomic Data Science Specialization
- Biology Meets Programming: Bioinformatics for Beginners
- Python for Genomic Data Science
- Bioinformatics Specialization
- Command Line Tools for Genomic Data Science
- Introduction to Genomic Technologies
References
- Sims D, Sudbery I, Ilott NE, Heger A, Ponting CP. Sequencing depth and coverage: key considerations in genomic analyses. Nature Reviews Genetics. 2014 Feb;15(2):121-32.
- Mamanova L, Coffey AJ, Scott CE, Kozarewa I, Turner EH, Kumar A, Howard E, Shendure J, Turner DJ. Target-enrichment strategies for next-generation sequencing. Nature methods. 2010 Feb;7(2):111-8.
- Making the Most of Your NGS Data: Understanding Metrics for Target-enriched NGS
- Bodi K, Perera AG, Adams PS, Bintzler D, Dewar K, Grove DS, Kieleczawa J, Lyons RH, Neubert TA, Noll AC, Singh S. Comparison of commercially available target enrichment methods for next-generation sequencing. Journal of biomolecular techniques: JBT. 2013 Jul;24(2):73.
- https://www.pacb.com/products-and-services/applications/whole-genome-sequencing/plant-animal/
- https://www.pacb.com/products-and-services/applications/whole-genome-sequencing/variant-detection/
This work is licensed under a Creative Commons Attribution 4.0 International License
Some of the links on this page may be affiliate links, which means we may get an affiliate commission on a valid purchase. The retailer will pay the commission at no additional cost to you.

