How to Determine Strandedness in RNA-seq Data (With Example)
Strandedness in RNA-seq
In order to analyze RNA-seq data, it is crucial to know whether the sequencing data was generated from stranded or unstranded libraries.
Because different RNA-seq tools rely on library preparation protocols, incorrect use of RNA-seq strandedness can lead to false positives in differential gene expression analysis and significant loss in mapping rates to reference genomes.
As an example, the htseq-count command (from HTSeq) requires
stranded parameter for counting the reads. By default, the stranded parameter is set to yes (stranded library). The
default stranded parameter of the htseq-count function could result in a significant loss of read counts if the
RNA-seq data is generated from an unstranded library.
Based on strandedness, the sequencing library could be fr-unstranded, fr-firststrand, and fr-secondstrand.
| Library | Description |
|---|---|
| fr-unstranded | Orientation of the original mRNA transcripts is not known read 1 (first sequenced read) and read 2 (second sequenced read) could be sequenced from either sense or antisense strand |
| fr-firststrand | Orientation of the original mRNA transcripts is known read 1 (first sequenced read) is sequenced from sense strand read 2 (second sequenced read) is sequenced from antisense strand |
| fr-secondstrand | Orientation of the original mRNA transcripts is known read 1 (first sequenced read) is sequenced from antisense strand read 2 (second sequenced read) is sequenced from sense strand |
Note: When reads are paired-end, read 1 is left read (R1) and read 2 is right read (R2). Single-end reads result in only one read (read 1).
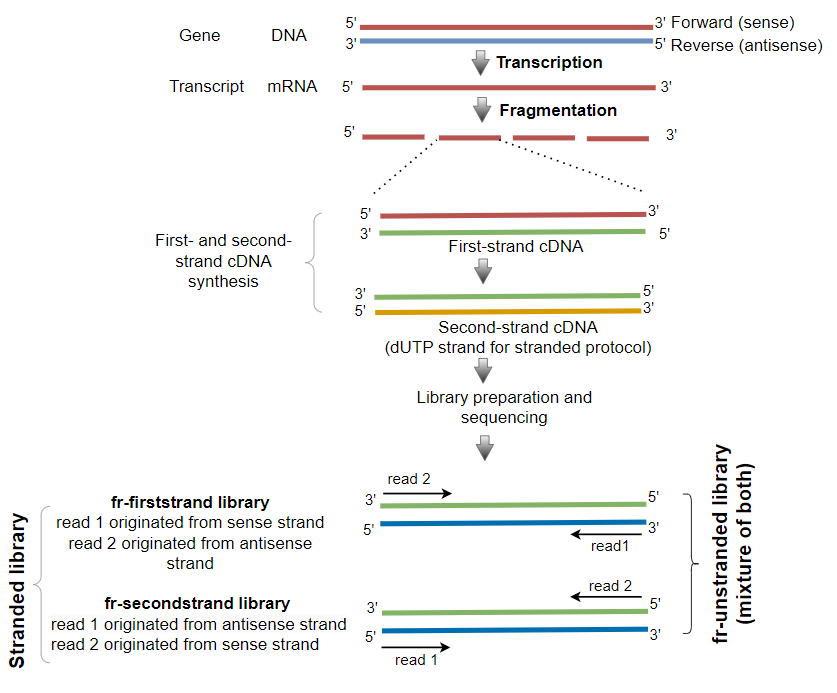
How to determine the strandedness of RNA-seq data?
The strandedness of RNA-seq data can be determined by comparing the strandedness of reads with that of orientation of the transcripts.
The infer_experiment.py script from the RSeQC package compares the
“strandedness of reads” with the known “strandedness of transcripts” to determine strandedness of RNA-seq data.
The “strandedness of transcripts” could be obtained from GTF file.
We will use the example of Arabidopsis thaliana RNA-seq data. You can download the genome sequence and gene annotation (GTF format) from Ensembl Plants.
The infer_experiment.py script requires a GTF file in BED format and an alignment file (SAM/BAM format).
Convert GTF to BED format using gtf2bed,
gtf2bed --gtf Arabidopsis_thaliana.TAIR10.56.gtf --bed Arabidopsis_thaliana.TAIR10.56.bed
You can use a STAR or HISAT2 aligner to obtain a alignment file in SAM/BAM format. Please read my articles on STAR and HISAT2 to obtain SAM/BAM file.
Now, run the infer_experiment.py script to determine the strandedness of RNA-seq data,
infer_experiment.py -i alignment.sam -r Arabidopsis_thaliana.TAIR10.56.bed
# output
Reading reference gene model Arabidopsis_thaliana.TAIR10.56.bed ... Done
Loading SAM/BAM file ... Total 200000 usable reads were sampled
This is PairEnd Data
Fraction of reads failed to determine: 0.0385
Fraction of reads explained by "1++,1--,2+-,2-+": 0.4824
Fraction of reads explained by "1+-,1-+,2++,2--": 0.4792
Based on the output obtained from infer_experiment.py, we can interpret it as below,
| Output | Interpretation |
|---|---|
| This is PairEnd Data | The RNA-seq data sequenced using paired-end sequencing |
| Fraction of reads failed to determine: 0.0385 | 3.85% reads were aligned to genome region where “strandedness of transcripts” could not be determined |
| Fraction of reads explained by “1++,1–,2+-,2-+”: 0.4824 | 48.24% of the reads could be explained by “1++,1–,2+-,2-+” (fr-secondstrand) |
| Fraction of reads explained by “1+-,1-+,2++,2–”: 0.4792 | 47.92% of the reads could be explained by “1+-,1-+,2++,2–” (fr-firststrand) |
A roughly equal number of reads are explained by the “1++,1-,2+-,2-+” and “1+-,1-+,2++,2-“. This concludes that RNA-seq data is unstranded.
If the RNA-seq data is stranded, you could get the majority of the reads explained by either “1++,1–,2+-,2-+” or “1+-,1-+,2++,2–”.
Tip: A majority of your reads are from “1+-,1-+,2++,2–”, which means they were sequenced using the fr-firststrand stranded method. When the majority of your reads come from “1++,1-,2+-,2-+”, the reads were sequenced with fr-secondstrand stranded methods.
Enhance your skills with courses on genomics and bioinformatics
- Genomic Data Science Specialization
- Biology Meets Programming: Bioinformatics for Beginners
- Python for Genomic Data Science
- Bioinformatics Specialization
- Command Line Tools for Genomic Data Science
- Introduction to Genomic Technologies
This work is licensed under a Creative Commons Attribution 4.0 International License
Some of the links on this page may be affiliate links, which means we may get an affiliate commission on a valid purchase. The retailer will pay the commission at no additional cost to you.

